Der primäre Immundefekt – Ein praktischer Leitfaden für den Kinderarzt

Einleitung
Es ist nicht einfach, im Verlaufe des ersten Lebensjahres, normale Infektionen von ungewöhnlichen zu unterscheiden. Das Immunsystem befindet sich in voller Entwicklung und Kinder sind einer grossen Anzahl Keime ausgesetzt, insbesondere bei kollektiver Betreuung. Ziel dieses Artikels ist es, die Elemente herauszuarbeiten, die an einen primären Immundefekt denken lassen, entsprechende Laborabklärungen vorzuschlagen und zu begründen, sowie einige Punkte hinsichtlich der Betreuung von Patienten mit einem Immundefekt darzulegen, da der Kinderarzt in der Erstversorgung eine zentrale Rolle einnimmt. Auf sekundäre Immundefekte, medikamentösen oder infektiösen Ursprungs, wird hier nicht eingegangen.
Es gibt zahlreiche, verschieden primäre Immundefekte. Sie können das angeborene sowie adaptative Immunsystem betreffen. Sie können transient (wie die Transitorische Hypogammaglobulinämie im Kindesalter) oder permanent vorhanden sein. Die Art der Infektion, die Geschwindigkeit, mit der sie sich ausbreitet, sowie weitere assoziierte allgemeine Symptome sind Schlüsselelemente bei der Identifizierung eines Immundefektes. Immundefekte werden heutzutage im Rahmen eines immunologischen Kontinuums klassifiziert, das nicht nur die erhöhte Infektanfälligkeit berücksichtigt, sondern auch das erhöhte Risiko einer Lymphoproliferation und Autoimmunität1,2). Das anfängliche klinische Erscheinungsbild ist sehr variabel und umfasst neben wiederholten, atypischen, langdauernden oder schweren Infekten, auch: Wachstumsstörungen, Zeichen einer Autoimmunität oder malignen Erkrankung. Der Praxispädiater nimmt bei der initialen Diagnosestellung sowie bei der langfristigen Betreuung von Patienten mit einem Immundefekt eine entscheidende Rolle ein: Dank seiner raschen Verfügbarkeit bei Infektionskrankheiten, der Koordination der verschiedenen beteiligten Fachärzte und der Beurteilung von psychosozialen Auswirkungen der Krankheit auf das Kind und seine Familie.
Bei welchem Kind muss ein Immundefekt vermutet werden
Es gibt wenig Daten zur normalen Anzahl Infektionen bei einem gesunden Kind während den ersten Lebensjahren. Die Zahlen variieren je nach Studie, mit einer Inzidenz von 3.7 bis 8 Atemwegsinfektionen jährlich bei Kindern im Vorschulalter3,4). Um das frühzeitige Erkennen von primären Immundefekten zu erleichtern, hat die Jeffrey Model Foundation5)1990 Warnzeichen ausgearbeitet. Diese Kriterien entsprechen denen der European Society for Immunodeficiencies (ESID)6) (Tabelle 1). Sie wurden als Instrument erarbeitet, um auf das Vorliegen eines Immundefekts bei Kindern aufmerksam zu machen und nicht als diagnostische Kriterien. In unserer Praxis ergänzen wir die Kriterien der Jeffrey Model Foundation durch zwei weitere: Das Auftreten von Infektionen mit üblicherweise harmlosen Keimen (z.B. Lymphadenitis durch atypische Mycobakterien) und Komplikationen durch abgeschwächte Lebendimpfstoffe7). Am meisten prädiktiv erweisen sich dabei das Vorkommen eines Immundefektes in der Familie, die Verabreichung intravenöser Antibiotika bei einer Sepsis und die Wachstumsstörung8). Einige Autoren haben seither weitere Kriterien vorgeschlagen, die mit einer grösseren Sensitivität einhergehen sollen9). Weiter können auch eine Autoimmunkrankheit oder eine Krebserkrankung erste Zeichen eines Immundefektes sein10).

Die Anzahl akuter Otitiden, die eine immunologische Abklärung veranlassen sollte, variiert von Land zu Land: Vier genügen gemäss der Jeffrey Model Foundation und der ESID. Die deutschen Empfehlungen setzen die Grenze bei sechs und die schweizerischen bei acht an5-7,9). Wichtig ist nicht nur die exakte Anzahl, sondern die Kombination von wiederholten Infektionen und weiteren Auffälligkeiten (Wachstumsstörung, chronische Durchfälle, usw.), die den Praxispädiater alarmieren sollten.
Kann ein humoraler Immundefekt die Klinik erklären, können die ersten Abklärungen beim Praxispädiater durchgeführt werden. In allen anderen Fällen empfehlen wir, den Patienten direkt an ein spezialisiertes Zentrum mit akkreditiertem Labor zu überweisen (insbesondere bei Verdacht auf einen Komplementdefekt, Defekt der Phagozytenfunktion, der T-Lymphozyten oder bei lebensbedrohlichen Infektionen).
Vorgehen
Der Verdacht auf einen Immundefekt erfordert eine ausführliche Anamnese. Dabei geht es insbesondere um die Schwangerschaft (Ultraschall, HIV-Risiko), die Zeit bis zum Abfall der Nabelschnur, Anzahl Infektionen, Wachstum und Entwicklung, chronischer Diarrhoe (Malabsorption? Lamblien?), Atopien (Asthma, Ekzeme), Autoimmunität, sowie um die Familienanamnese (schwere, langdauernde, ungewöhnliche oder wiederholte Infektionskrankheiten, frühe oder unerklärte Todesfälle in der Familie, Konsanguinität).

Ein kompletter Status ist notwendig mit speziellem Augenmerk auf das Vorhandensein der Tonsillen, einer Hepatosplenomegalie oder Lymphadenopathie. Ebenso sollen das Integument, die Zähne und Haare sorgfältig untersucht werden (Ekzeme, Livedo, partieller Albinismus, Teleangiektasien, Nagelveränderungen, Form der Zähne, brüchige und spärliche Haare).
Erste Laborabklärungen umfassen ein vollständiges Blutbild zur Erfassung von Zytopenien, insbesondere Neutropenie oder Lymphopenie, eine gewichtsadaptierte Bestimmung der Immunglobuline (IgA, IgG und IgM) und Impfantwort. Als Standard gelten die Impfantikörper auf Tetanus, Diphtherie und Pneumokokken. Die beiden ersteren wiederspiegeln die Fähigkeit, auf Proteinantigene zu reagieren, was die Zusammenarbeit der T- und B-Lymphozyten voraussetzt. Die Bestimmung der Impfantwort auf die 23 Pneumokokkenserotypen erlaubt einerseits die Immunantwort auf die in Prevenar13 enthaltenen Serotypen zu bestimmen, andererseits auch die natürliche Reaktion auf Polysaccharid-Antigene, sowie eine gezielte Immunantwort auf Polysaccharide nach Verabreichung von Pneumovax23. Die Antwort auf Polysaccharide benötigt ausschliesslich B-Lymphozyten, sie führt nicht zur Bildung von B-Gedächtniszellen und ist bis zum Alter von zwei Jahren physiologisch unzureichend. Wird ein T-Lymphozytendefekt vermutet, wird in einem anerkannten Immunologielabor eine Untersuchung der Lymphozytensubgruppen durchgeführt, in gewissen Fällen auch der Lymphozytenproliferation (die T-Lymphozyten werden mit gewissen, aus Pathogenen gewonnene Antigenen in Kontakt gebracht und die darauffolgende Anzahl Zellteilungen gezählt). Wird ein Phagozytosedefekt vermutet, kann die Granulozytenfunktion durch den Dihydrorhodamin-Oxydationstest geprüft werden. Komplementbestimmungen werden in der pädiatrischen Poliklinik für Immunologie bezüglich dem klassischen, dem alternativen sowie dem Lektin-Weg bestimmt, u.U. auch dem terminalen Endkomplex. Je nach Ergebnis werden ergänzende Untersuchungen durchgeführt.
Welche Klinik – welcher Immundefekt
Die Klassifizierung der primären Immundefekte gründet auf dem defekten immunologischen Mechanismus und den vorherrschenden klinischen Zeichen. Die Art der Infektion und die damit zusammenhängenden Symptome weisen auf den möglichen Defekt hin (Abb. 1). Wiederholte bakterielle ORL- und Lungeninfekte lassen eher an einen humoralen Defekt (der Immunglobuline produzierenden B-Lymphozyten) denken; wiederholte virale Infekte hingegen, z.B. atypische Mycobakterien, komplizierte Varizellen, eine Pneumocystis jirovecii-Pneumonie hingegen an einen zellulären (T-Lymphozyten) Defekt. Rezidivierende Hautabszesse mit Granulomen weisen auf einen Phagozytosedefekt hin und Infektionen mit eingekapselten Keimen oder eine Meningokokken-Meningitis auf einen Komplementdefekt1).
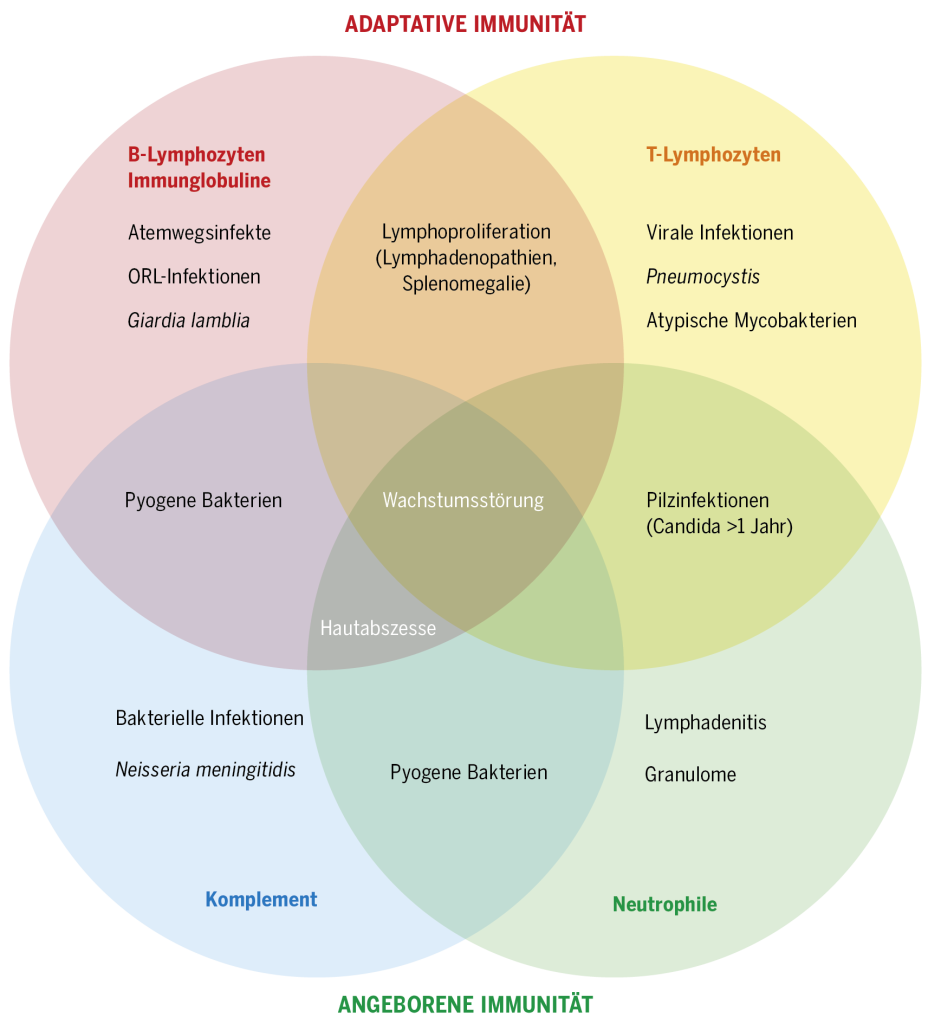
Defekte der adaptativen Immunität
Humoraler Defekt (B-Lymphozyten)
Antikörpermangel im Zusammenhang mit einer Dysfunktion der B-Lymphozyten sind die häufigsten (ca. 50%) primären Immundefekte10). Es sollen hier die wichtigsten dieser Defekte beschrieben werden, die sich meist durch bakterielle ORL- und Atemwegsinfekte auszeichnen.
1. Selektiver IgA-Mangel
IgA-Mangel wird regelmässig durch Praxispädiater festgestellt, insbesondere beim Zöliakie-Screening. Es handelt sich um den häufigsten Immundefekt beim Menschen, wobei die Prävalenz von der ethnischen Herkunft abhängig ist: in Saudi-Arabien 1:14211), in Japan 1:14’84012). Die Diagnose kann bei Kindern über 4 Jahren mit erhöhter Infektanfälligkeit, Autoimmunerkrankung oder positiver Familienanamnese und nicht messbarem IgA (zweimal gemessen) gestellt werden. Dabei müssen die IgG-, IgM-Werte und die Impfantwort normal sein, sowie Ursachen einer sekundären Hypogammaglobulinämie und ein assoziierter T-Zelldefekt ausgeschlossen werden13). Aus Beobachtungsstudien weiss man, dass bis zu 2/3 der Patienten asymptomatisch sind14). Die symptomatischen Patienten leiden an rezidivierenden Sinus- und Lungeninfektionen, autoimmunen (vor allem Zöliakie) und atopischen Erkrankungen, Magendarmerkrankungen und chronischer Lambliasis. Die IgG- und IgM-Werte sollten bei allen Patienten bestimmt werden, bei welchen ein nicht messbarer IgA-Werte zufällig festgestellt wurde. Besteht einzig ein IgA-Mangel und leidet der Patient weder an rezidivierenden Infektionen (oder anderen Red Flags) noch Zeichen einer Autoimmunität und ist die Familienanamnese negativ, können die Nachkontrollen durch den behandelnden Arzt durchgeführt werden. Es besteht kein Konsens über Empfehlungen zu Nachkontrollen, insbesondere darüber, ob wiederholte Immunglobulinmessungen in regelmäßigen Abständen sinnvoll sind. Besondere Aufmerksamkeit muss Kindern mit häufigen Infekten oder Anzeichen einer Autoimmunkrankheit geschenkt werden.
2. Transitorische Hypogammaglobulinämie des Kindesalters
Die Inzidenz der transitorischen Hypogammaglobulinämie des Kindesalters liegt zwischen 0.06 und 1.1/1000 Geburten15). Sie wird definiert durch einen zweimal unter der Altersnorm liegenden IgG-Wert im Verlauf der ersten drei Lebensjahre, dem Ausschluss anderer Ursachen einer Hypogammaglobulinämie und der spontanen Resolution vor dem Alter von vier Jahren13). Die Diagnose wird deshalb immer a posteriori gestellt, nach der Normalisierung der IgG-Werte. Bei den meisten Patienten bestehen wiederholte oder schwere Infekte der oberen und unteren Luftwege, sowie eine Atopie in Form von Nahrungsmittelallergien, Asthma oder Ekzem16). Die Phänotypisierung der Lymphozyten fällt normal aus und die Impfantworten sind im Allgemeinen adäquat oder normalisieren sich mit dem Beheben der Hypogammaglobulinämie17). Je nach Häufigkeit und Schwere der Infektionen kann eine Antibiotikaprophylaxe oder eine Immunglobulinsubstitution vorgeschlagen werden18).
3. Allgemeine variable Immundefizienz (CVID)
Die CVID ist ein häufiger Antikörpermangel, mit einer geschätzten europäischen Prävalenz von 1:25’00019). Sie stellt eine heterogene Gruppe von Defiziten in der Antikörperproduktion dar, die sich durch häufige Infektionen (Bronchitiden, Sinusitiden, Otitiden, Pneumonien, Magendarminfektionen), entzündliche Lungenerkrankungen (mit interstitiellen Folgeschäden), autoimmune Störungen (autoimmune Zytopenien, Störung der Schilddrüsenfunktion), Wachstumsstörung, Enteropathien und erhöhtem Risiko einer Lymphoproliferation (insbesondere Non-Hodgkin-Lymphom und Magendarmtumoren) auszeichnet19). Die Diagnose kann bei über 4jährigen Patienten mit vermehrter Infektanfälligkeit oder autoimmunen Störungen, mindestens zweimal gemessenen IgG- und IgA-Werten (+/- IgM) unter der Altersnorm und ungenügender Impfantwort gestellt werden, bei denen kein T-Lymphozytendefekt besteht und die Ursachen einer sekundären Hypogammaglobulinämie (Medikamente, renaler oder gastrointestinaler Verlust) ausgeschlossen wurden13). Es handelt sich um eine meist während dem 2.-3. Lebensjahrzehnt, jedoch in 25% der Fälle bereits im Kindes- oder Jugendalter gestellte Diagnose20). Die Frage ob es sich bei der CVID-Diagnose im Kindes- bzw. Erwachsenenalter um verschiedene nosologische Entitäten handelt, bleibt umstritten19). Die Behandlung beruht auf Immunglobulinsubstitutionen, Antibiotikaprophylaxe und Früherkennung von Komplikationen.
4. Weitere seltenere humorale Defekte
Die Agammaglobulinämie gehört den selteneren und schwerer verlaufenden humoralen Defekten an. Sie zeichnet sich durch das Fehlen von B-Lymphozyten aus. Die häufigste Form ist die X-chromosomale Agammaglobulinämie (Bruton-Krankheit), die 1 Knabe auf 190’000 trifft. Oft treten Infektionen beim Säugling schon ab dem 3. Lebensmonat (mit dem Verschwinden der mütterlichen IgG) auf, im Allgemeinen vor dem Alter von 5 Jahren21). Die Krankheit muss bei einem männlichen Säugling ohne Tonsillen, sehr tiefen IgG-, IgA- und IgM-Werten und einer stark erniedrigten B-Lymphozytenzahl (<1-2% der Gesamt-Lymphozytenzahl) erwogen werden.
Zellulärer und kombinierter Defekt (T- und B-Lymphozyten)
Bei Patienten mit der T-Zell-Defekten besteht auf Grund der fehlenden, für die Produktion spezifischer Antikörper notwendigen T- und B- Zell Zusammenarbeit, oft auch eine Störung der B-Zell-Immunität. Die schwerste Form, der schwere kombinierte Immundefekt (SCID) ist mit einer geschätzten Inzidenz von 1:50’000 selten, während schwer verlaufende T-Lymphopenien anderer Ätiologie eine geschätzte Inzidenz von 1:10’000 haben22). Die Diagnose SCID ist ein pädiatrischer Notfall; die frühzeitige Behandlung durch Transplantation hämatopoetischer Stammzellen vor dem Auftreten von möglicherweise letalen Infektionen weist eine hohe Erfolgsrate auf23). Aus diesem Grund werden die schweren T- und B-Zell-Immundefekte in der Schweiz seit 2019 durch das Neugeborenenscreening erfasst24). Bei fehlender Früherkennung weisen die Kinder ab dem Alter von 3-4 Monaten schwere und häufige Infektionen mit opportunistischen Keimen auf, insbesondere mit Pilzen und Viren (Candida albicans, CMV, Pneumocystis jirovecii), sowie chronische Durchfälle, Wachstumsstörung und neonatale Hautausschläge1). Die T-Zell oder T-und B-Zell-Defekte können mit Missbildungssyndromen einhergehen, wie das DiGeorge-Syndrom (mit Thymus Hypo- oder Aplasie) oder die Ataxia teleangiectatica. Wichtig scheint uns, daran zu erinnern, dass bei Diagnose eines T-Lymphozytendefekts, insbesondere CD4+, eine HIV-Infektion ausgeschlossen werden muss.
Defekt der angeborenen Immunität
Defekte der Neutrophilen
Die Defekte der Neutrophilen können quantitativ, wie bei der zyklischen Neutropenie, qualitativ, wie bei der chronischen Granulomatose, oder beides sein, wie bei der schweren kongenitalen Neutropenie. Ob quantitativ oder qualitativ, das Resultat ist ähnlich und besteht in einer erhöhten Anfälligkeit für schwere Pilz- (z.B Candida albicans, Aspergillus) und bakterielle, v.a. pyogene (z.B. Staphylococcus aureus, Pseudomonas aeruginosa) Infektionen der Haut, der Lymphknoten und der Atemwege1,25). Das Infektionsrisiko ist bei weniger als 200 Neutrophilen/mm3 erhöht, bei 200-1000 Neutrophilen/mm3 mässig und bei mehr als 1000 Neutrophilen/mm3 gering26). Typischerweise besteht keine erhöhte Anfälligkeit für virale Infekte. Weitere häufig assoziierte Krankheitszeichen sind eine verzögerte Narbenbildung, Ekzeme, Stomatitis und Wachstumsstörung. Als Laborbefunde findet man eine Neutropenie, einen abnormen DHR-Test und häufig persistierende Entzündungsparameter.
Komplementdefekte
Komplementdefekte sind selten und stellen weniger als 1% der primären Immundefekte dar1,27). Die Defekte des klassischen Weges (C1q, C2, C4) prädisponieren, zusätzlich zum erhöhten Risiko bakterieller Infektionen, zu Autoimmunkrankheiten, insbesondere zu einem Systemischen Lupus Erythematodes28). C3-Defekte gehen mit häufigen schweren Infektionen mit eingekapselten Bakterien, insbesondere Streptococcus pneumoniae und Haemophilus influenzae einher29). Bei Defekten der terminalen Endstrecke (Membranangriffskomplex) sowie des alternativen Weges, insbesondere des Properdins, besteht eine starke Neigung zu Neisseria meningitidis-Infektionen29). Die Behandlung umfasst gezielte Impfungen gegen Pneumokokken, Haemophilus und Meningokokken, und eine frühzeitige antibiotische Therapie bei Infektionen.
Weitere Defekte der angeborenen Immunität
Defekte der NK-Zellen sind sehr selten. Sie zeichnen sich durch schwere und langdauernde Infektionen mit Viren der Herpesfamilie aus (HSV, EBV, CMV, VZV), sowie einer vermehrten Neigung, Tumoren und eine Autoimmunität zu entwickeln 30).
Die Rolle des Pädiaters bei der Betreuung von Patienten mit einem primären Immundefekt
Die Rolle des Pädiaters ist bei der Betreuung von Patienten mit einem primären Immundefekt wichtig, um die notwendigen Impfungen durchzuführen und bei Infektionen und Epidemien eingreifen zu können, um proaktiv Wachstum und Entwicklung zu überwachen, und die psychosozialen Auswirkungen zu erfassen. Genauere Angaben zu den wichtigsten Punkten sind in Tabelle 3 zusammengefasst.

Schlussfolgerung
Die Betreuung von Patienten mit einem Immundefekt ist komplex. In vielen Fällen sind sie nicht nur anfälliger für Infektionen, sondern haben auch ein erhöhtes Risiko für Autoimmun-, atopische und/oder Krebskrankheiten. Die Warnsignale sind eine Hilfe für den Praxispädiater und die Bevölkerung, um Kinder mit einem Immundefektrisiko zu erkennen, es sind hingegen keine strikten diagnostischen Kriterien. Es ist in der Tat nicht die Anzahl der Mittelohrentzündungen, die den Verdacht des Kinderarztes auf einen Immundefekt wecken soll, es ist das Aufeinandertreffen verschiedener Symptome, was ihn dazu bringen soll, Anamnese und klinische Untersuchung zu vertiefen, und erste Laborabklärungen zu veranlassen.
Gewisse Patienten mit einem Immundefekt werden regelmässig von Fachärzten gesehen – die Rolle des Pädiaters bleibt zentral, um die Informationen zusammenzufassen, um bei einer Infektion oder bei Epidemien rasch für den Patienten verfügbar zu sein, um sich zu vergewissern, dass die Medikamente korrekt eingenommen werden, um die notwendigen Impfungen vorzunehmen und schliesslich, um die Auswirkungen der Krankheit auf Wachstum, Entwicklung und psychosoziale Situation zu erfassen.
Referenzen
- McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy, Asthma Clin Immunol [Internet]. 2018;14(s2):1–12. Available from: https://doi.org/10.1186/s13223-018-0290-5
- Savic S, Caseley EA, McDermott MF. Moving towards a systems-based classification of innate immune-mediated diseases. Nat Rev Rheumatol [Internet]. 2020;16(4):222–37. Available from: http://dx.doi.org/10.1038/s41584-020-0377-5
- JORDAN WS, DENNY FW, BADGER GF, CURTISS C, DINGLE JH, OSEASOHN R, et al. a Study of Illness in a Group of Cleveland Families. Am J Epidemiol. 1958;68(2):190–212.
- Grüber C, Keil T, Kulig M, Roll S, Wahn U, Wahn V, et al. History of respiratory infections in the first 12 yr among children from a birth cohort. Pediatr Allergy Immunol. 2008;19(6):505–12.
- Foundation JM. https://www.info4pi.org/library/educational-materials/10-warning-signs.
- European Society for Immunodeficiencies. https://esid.org/Working-Parties/Clinical-Working-Party/Resources/10-Warning-Signs-of-PID-General.
- Déficience immunitaire Suisse. https://www.deficience-immunitaire-suisse.ch/fr/signaux-alarme/-chez-enfants/?oid=1872&lang=fr.
- Subbarayan A, Colarusso G, Hughes SM, Gennery AR, Slatter M, Cant AJ, et al. Clinical features that identify children with primary immunodeficiency diseases. Pediatrics. 2011;127(5):810–6.
- Lankisch P, Schiffner J, Ghosh S, Babor F, Borkhardt A, Laws HJ. The Duesseldorf Warning Signs for Primary Immunodeficiency: Is it Time to Change the Rules? J Clin Immunol. 2015;35(3):273–9.
- Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol [Internet]. 2014;136(5):1186-1205.e78. Available from: http://dx.doi.org/10.1016/j.jaci.2015.04.049
- Al-Attas RA, Rahi AHS. Primary antibody deficiency in Arabs: First report from Eastern Saudi Arabia. J Clin Immunol [Internet]. 1998 [cited 2022 Jan 24];18(5):368–71. Available from: https://pubmed.ncbi.nlm.nih.gov/9793829/
- Kanoh T, Mizumoto T, Yasuda N, Koya M, Ohno Y, Uchino H, et al. Selective IgA Deficiency in Japanese Blood Donors: Frequency and Statistical Analysis. Vox Sang. 1986;50(2):81–6.
- ESID. ESID Registry – Working definitions for clinical diagnosis of PID [Internet]. ESID Registry – Working definitions for clinical diagnosis of PID. 2019. Available from: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria
- Aytekin C, Tuygun N, Gokce S, Dogu F, Ikinciogullari A. Selective IgA deficiency: Clinical and laboratory features of 118 children in Turkey. J Clin Immunol. 2012;32(5):961–6.
- Tiller TL, Buckley RH. Transient hypogammaglobulinemia of infancy: Review of the literature, clinical and immunologic features of 11 new cases, and long-term follow-up. J Pediatr [Internet]. 1978 [cited 2022 Jan 24];92(3):347–53. Available from: https://pubmed.ncbi.nlm.nih.gov/632973/
- Moschese V, Graziani S, Avanzini MA, Carsetti R, Marconi M, La Rocca M, et al. A prospective study on children with initial diagnosis of transient hypogammaglobulinemia of infancy: Results from the Italian Primary Immunodeficiency Network. Int J Immunopathol Pharmacol. 2008;21(2):343–52.
- Dorsey MJ, Orange JS. Impaired specific antibody response and increased B-cell population in transient hypogammaglobulinemia of infancy. Ann Allergy, Asthma Immunol [Internet]. 2006;97(5):590–5. Available from: http://dx.doi.org/10.1016/S1081-1206(10)61085-X
- Enders FB, Conti F, Candotti F, Angelini TF. Hypogammaglobulinémie transitoire de l’enfant. Rev Med Suisse. 2017;13(557):739–42.
- Gathmann B, Mahlaoui N, Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1).
- Urschel S, Kayikci L, Wintergerst U, Notheis G, Jansson A, Belohradsky BH. Common Variable Immunodeficiency Disorders in Children: Delayed Diagnosis Despite Typical Clinical Presentation. J Pediatr. 2009;154(6):888–94.
- Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: Report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85(4):193–202.
- van der Burg M, Mahlaoui N, Gaspar HB, Pai SY. Universal Newborn Screening for Severe Combined Immunodeficiency (SCID). Front Pediatr. 2019;7(September):1–5.
- Pai S-Y, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation Outcomes for Severe Combined Immunodeficiency, 2000–2009. N Engl J Med [Internet]. 2014 Jul 31 [cited 2022 Jan 24];371(5):434–46. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1401177
- Trück J, Prader S, Natalucci G, Hagmann C, Brotschi B, Kelly J, et al. Swiss newborn screening for severe T and B cell deficiency with a combined TREC/KREC assay – Management recommendations. Swiss Med Wkly. 2020;150(25–26):1–6.
- Dinauer MC. Disorders of neutrophil function: An overview. Methods Mol Biol [Internet]. 2014 [cited 2022 Jan 24];1124:501–15. Available from: https://pubmed.ncbi.nlm.nih.gov/24504971/
- Donadieu J, Fenneteau O. Constitutional and acquired neutropenia. Vol. 2, EMC – Hematologie. Elsevier Masson SAS; 2005. p. 158–86.
- Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35(8):696–726.
- Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front Immunol. 2016;7(FEB):1–22.
- Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23(4):740–80.
- Orange JS, Ballas ZK. Natural killer cells in human health and disease. Clin Immunol. 2006;118(1):1–10.
Weitere Informationen
Autor:innen
-
Dr med. Tiphaine ArlabosseUnité d’immunologie, allergologie et rhumatologie pédiatrique, Service de Pédiatrie, Département Femme-Mère-Enfant, CHUV, Lausanne
-
Dr med. Katerina TheodoropoulouUnité d’immunologie, allergologie et rhumatologie pédiatrique, Service de Pédiatrie, Département Femme-Mère-Enfant, CHUV, Lausanne
-
Prof. Dr med. Fabio CandottiService d’immunologie et allergie, CHUV et UNIL, Lausanne