
Introduction
L’obésité dite « commune » est fortement influencée par des facteurs sociaux, environnementaux et génétiques. Les études familiales, chez les jumeaux et des enfants adoptés ont montré que 40-70% du poids corporel s’explique par la variation génétique dans la population(1).
En revanche, les obésités monogénique et syndromique sont des formes d’obésité sévère et précoce qui s’accompagnent souvent d’une hyperphagie et qui ne sont que très peu influencées par l’environnement de l’enfant. Les gènes responsables de ces rares formes d’obésité sévère appartiennent à la voie de la leptine-melanocortine hypothalamique, qui régule la satiété et le poids corporel(2). Le diagnostic précoce des obésités monogéniques et syndromiques est important pour détecter et suivre les anomalies et comorbidités associés, prévenir la stigmatisation des patient·e·s et parents, donner un conseil génétique et, si indiqué, mettre en place un traitement pharmacologique spécifique(3).
Patiente
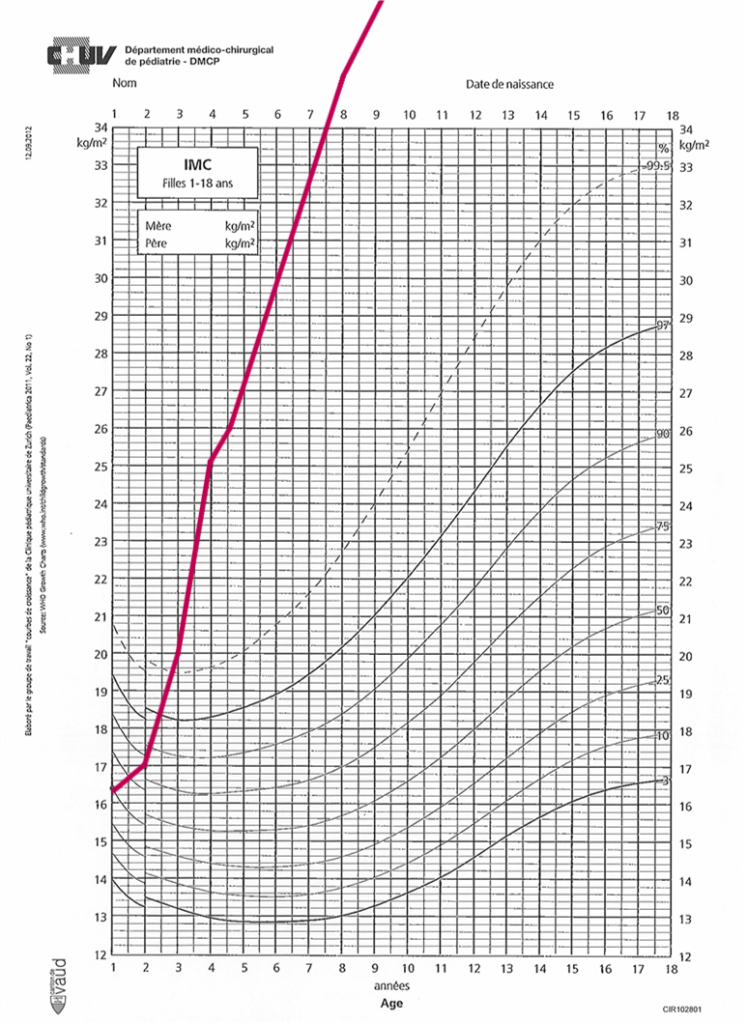
Laura est une fille qui se présente à l’âge de 5 ans avec une obésité sévère, IMC à + 6.6 DS. Elle est née avec un poids de naissance dans les normes et l’obésité s’est progressivement développée dès l’âge de 18 mois et s’accompagne d’une hyperphagie. Laura recherche activement la nourriture, réclame fréquemment à manger et a une frustration importante quand la mère essaie de restreindre la nourriture. Elle a autrement un développement normal et aucun antécédant de santé notable. Laura a une insulino-résistance importante (index HOMA 7.7) déjà à 5 ans. Malgré un suivi multidisciplinaire intensif et sa participation aux groupes thérapeutiques, son obésité reste très sévère et à 12 ans elle à un IMC à 49.71kg/m2 (+5.42 DS).
Les 2 parents de Laura présentent une obésité. La mère a eu récemment un bypass gastrique. Le père a perdu du poids avec des analogues de GLP-1.
Des analyses génétiques faites dans le contexte d’une obésité précoce et très sévère, accompagnée d’une hyperphagie révèlent une mutation hétérozygote du MC4R héritée de sa mère. Elle est mise sous traitement par analogues de GLP-1 avec une bonne réponse.
Physiologie de la régulation du poids corporel
La régulation du poids corporel repose sur un équilibre subtil entre des processus homéostatiques et cognitivo-émotionnels, qui impliquent une interaction multifactorielle entre des hormones et des substances messagères au sein de circuits de régulation complexes. Une perturbation de ces systèmes peut entraîner un déséquilibre entre l’apport et la dépense énergétique, conduisant à l’obésité, ou au contraire à une prise de poids insuffisante(4,5). Dans les conditions de vie modernes, favorisant les prédispositions sous-jacentes à l’obésité, la prise pondérale est bien plus facile à atteindre que la perte de poids, en raison des mécanismes régulateurs intrinsèques(5,6).
Systèmes cérébraux impliqués dans la régulation du poids corporel
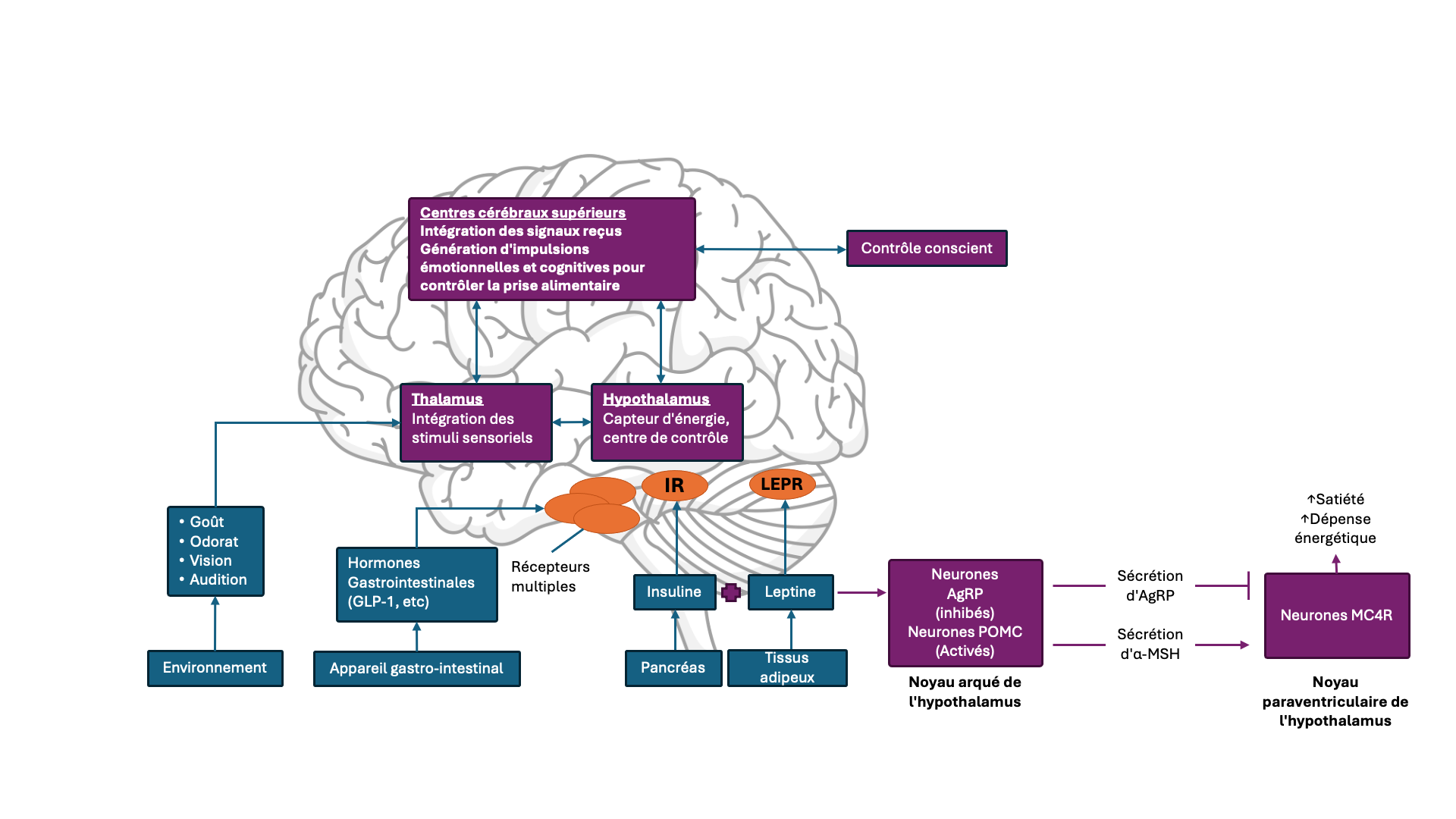
1. Homéostasie énergétique : Ce système est régulé principalement par des centres cérébraux situés dans l’hypothalamus et le tronc cérébral, et agit de manière inconsciente. Il intègre des signaux à long terme provenant des réserves énergétiques du tissu adipeux (par exemple la leptine), ainsi que des signaux à court terme, liés à la faim et à la satiété, issus du tractus gastro-intestinal. Un bilan énergétique négatif, entraînant une réduction de la masse grasse, diminue les niveaux de leptine, ce qui augmente la sensation de faim. À l’inverse, la distension gastrique et la libération d’hormones gastro-intestinales, telles que l’insuline induisent temporairement une sensation de satiété(4,5,7).
2. Contrôle cognitivo-émotionnel : Ce système, régulé par les centres cérébraux supérieurs, agit de manière consciente. Il combine les signaux homéostatiques avec des stimuli externes (p.ex., l’odeur ou le goût des aliments), des expériences personnelles et des émotions. Les perturbations de ce système peuvent se manifester par une alimentation émotionnelle ou des troubles du comportement alimentaire. Les aliments riches en calories activent davantage les zones de récompense cérébrales chez les personnes en excès pondéral que chez celles de poids normal(4,5,7).
Ces deux systèmes interagissent étroitement, le contrôle cognitivo-émotionnel étant fortement influencé par les circuits homéostatiques. Des perturbations dues à des facteurs génétiques, à des pathologies, à des lésions traumatiques touchant les centres régulateurs, ou à une programmation prénatale peuvent modifier le seuil de régulation du poids corporel, rendant inefficace toute tentative consciente de contrôle des apports alimentaires(5).
La voie leptine-mélanocortine
Cette voie constitue un mécanisme effecteur clé de la signalisation induite par la leptine au niveau cérébral. Le noyau arqué (ARC), situé à la base de l’hypothalamus, est le site principal d’action centrale de la leptine(8,9), hormone peptidique principalement sécrétée par le tissu adipeux en lien avec la quantité du tissu adipeux et au bilan énergétique. L’ARC de l’hypothalamus héberge les neurones NPY/AgRP (Neuropeptide Y/Agouti Related Peptide) et Proopiomélanocortine (POMC), deux populations neuronales distinctes et antagonistes. Ainsi, des niveaux élevés de leptine activent les neurones POMC et inhibent les neurones AgRP/NPY. Le POMC, un précurseur hormonal, est clivé en différents peptides tels que l’hormone α-mélanotrope (α-MSH), grâce aux enzymes Proprotein convertase subtilisin/kexin-type 1 (PCSK1) et Carboxypeptidase (CPE), entre autres(10). L’ α-MSH active les récepteurs de la mélanocortine-4 (MC4R) situés dans le noyau paraventriculaire de l’hypothalamus, induisant une sensation de satiété et une diminution de la prise alimentaire(5,11) et une augmentation de la dépense énergétique. .
À l’inverse, des niveaux faibles de leptine activent les neurones AgRP/NPY et inhibent les neurones POMC, induisant la sensation de faim et la prise alimentaire(5).
La liaison du facteur neurotrophique dérivé du cerveau (BDNF) à son récepteur, le récepteur tyrosine kinase neurotrophique 2 (NTRK2), est considéré comme un effecteur de la plasticité synaptique médiée par la leptine. Le facteur de transcription SIM1 (single-minded family basic helix-loop-helix [bHLH] transcription factor 1) est essentiel au développement du PVN(11).
Ce système joue un rôle central dans la pathogénèse de l’obésité et est une cible pour des thérapies spécifiques. Les mutations des gènes impliqués dans la voie leptine-mélanocortine-MC4R, telles que le LEP, LEPR, POMC, PCSK1, CPE, MC4R, BDNF, NTRK2 et SIM1 sont associées à une hyperphagie et une obésité sévère et d’apparition précoce(11).
Caractéristiques cliniques de l’obésité monogénique et syndromique
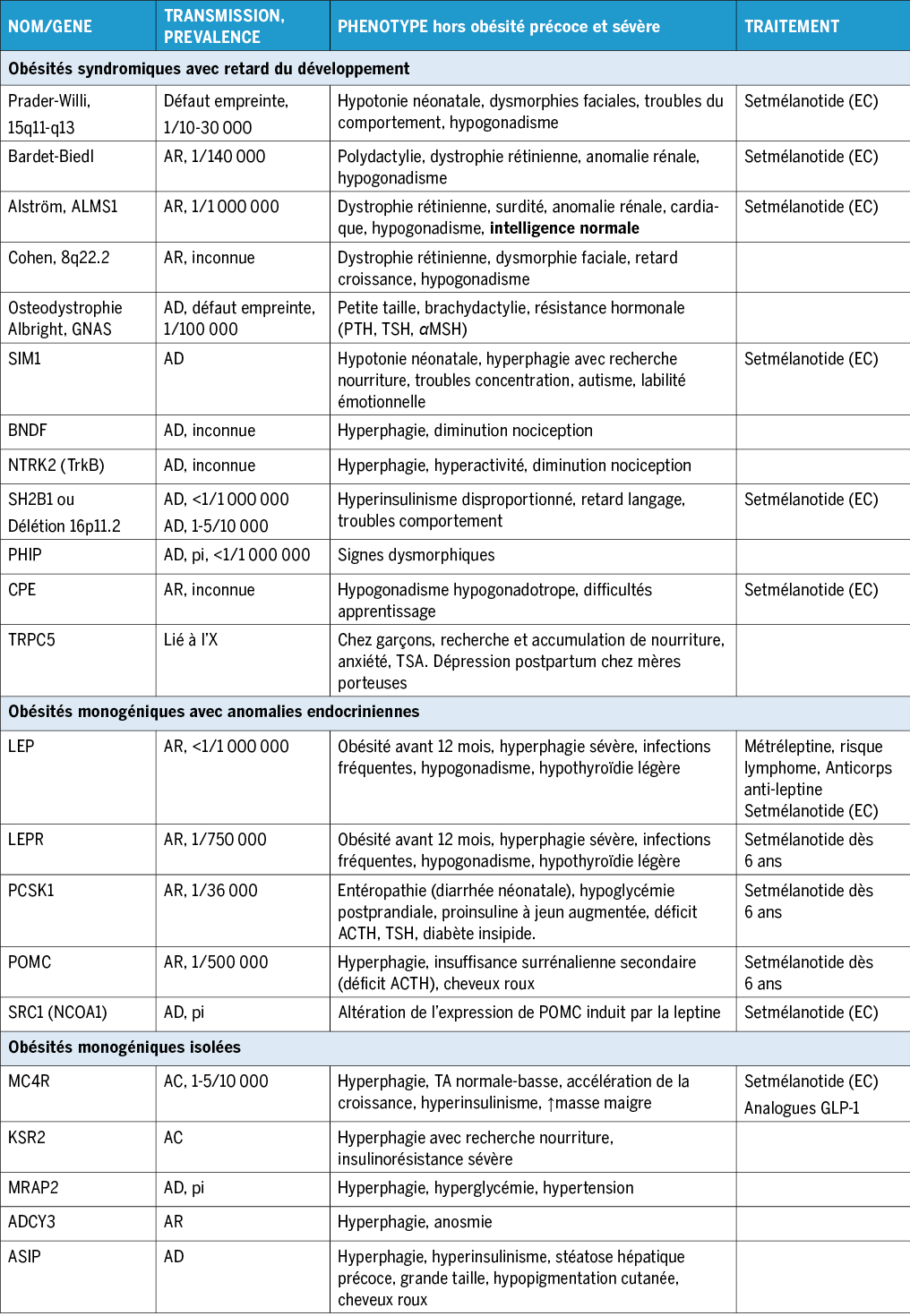
Les enfants atteints d’une obésité monogénique et syndromique ont une prise de poids rapide depuis la naissance et développent une obésité sévère avant l’âge de 5 ans. La prise de poids excessive est en lien avec la présence d’une hyperphagie, qui implique une consommation disproportionnée d’aliments, un manque de satiété, une préoccupation excessive pour la nourriture, une recherche et accumulation d’aliments et une détresse importante en cas de refus de nourriture(1). Cependant, reconnaitre et mesurer l’hyperphagie est difficile et son intensité dépend de la cause génétique sous-jacente, étant plus sévère et précoce dans le déficit en leptine ou mutation du récepteur de la leptine ainsi que dans le syndrome de Bardet-Bidel. Le questionnaire de Dykens est un outil qui a montré son utilité pour aider à évaluer le comportement hyperphagique dans d’autres obésités syndromiques et monogéniques(12).
Des caractéristiques spécifiques peuvent nous aider à orienter plus précisément notre suspicion clinique. Le retard du développement, les difficultés d’apprentissage et les troubles du comportement orientent vers une étiologie syndromique de l’obésité(13). Au contraire, la présence d’une obésité sévère isolée ou des altérations endocriniennes associées suggèrent une obésité monogénique(14).
Ainsi, le déficit en leptine ou les mutations du récepteur de la leptine, maladies extrêmement rares, s’associent à un hypogonadisme hypogonadotrope et une hypothyroïdie légère ainsi qu’a des infections fréquentes dans les formes homozygotes. Le poids de naissance est normal et l’hyperphagie sévère, avec en général un IMC > 27 kg/m2 à 2 ans et >33 kg/m2 à 5 ans(15).
Les mutations du gène MC4R sont trouvées dans 2 à 6 % des cas d’obésité extrême chez l’enfant et sont la cause la plus fréquente d’obésité monogénique. Les formes homozygotes sont associées à une hyperphagie plus sévère et un IMC plus élevé que les formes hétérozygotes. Les patient·e·s ont en outre une accélération de la croissance et une tension artérielle basse pour le dégrée d’obésité(2).
Le déficit en POMC se manifeste par une obésité avec hyperphagie, résultat du manque de α-MSH, ainsi qu’un déficit précoce en ACTH qui produit une insuffisance surrénalienne. Les cheveux roux et la peau claire résultent d’un manque de stimulation du récepteur MC1R des mélanocytes(16).
Le déficit en PCSK1 provoque une diminution du clivage des certaines prohormones (POMC, GHRH, THRH, GHRH, provasopressine, proglucagon et proinsuline) ce qui peut provoquer une obésité ainsi que des déficits hormonaux. L’altération du clivage des peptides gastriques, entraîne une malabsorption néonatale sévère(17).
La cause la plus fréquente d’obésité syndromique est le syndrome de Prader-Willi (SPW), qui se caractérise par une hypotonie néonatale sévère et des difficultés alimentaires suivies d’une hyperphagie sévère et du développement d’une obésité. En général, le SPW inclue une dysmorphie faciale, une déficience intellectuelle et des troubles du comportement, ainsi que souvent des perturbations endocriniennes et un retard de croissance, secondaires à un dysfonctionnement de l’hypothalamus, et une augmentation de l’incidence du diabète de type 2(17). L’apnée obstructive du sommeil (AOS) est fréquente chez les patient·e·s atteint·e·s du SPW, en particulier après l’âge de 2 ans, et est très répandue chez les adolescent·e·s et les adultes concernant jusqu’à 80 %.
Le syndrome de Bardet-Biedel (BBS) est une ciliopathie caractérisée par une polydactylie post-axiale à la naissance, une obésité sévère, une dystrophie rétinienne, un retard du développement, des anomalies rénales et un hypogonadisme. Les symptômes oculaires de manifestent vers l’âge de 8 ans(18). Dans le syndrome d’Alström (SA), aussi une ciliopathie avec atteinte multi-systémique, les troubles de la vision apparaissent plus précocement et il n’y a pas de déficit cognitif associé. L’ostéodystrophie héréditaire d’Albright (AHO) se caractérise par une obésité précoce, un retard du développement, un visage arrondi, une petite taille, une brachydactylie, et des ossifications sous-cutanées, toutes symptômes étant de pénétrance variable. Dans les variants d’origine maternelle s’ajoute une résistance hormonale principalement à la parathormone (PTH) et à la thyréostimuline (TSH)(16).
Diagnostic des obésités monogéniques et syndromiques
L’« Endocrine Society » recommande une évaluation génétique à la recherche d’une obésité monogénique ou syndromique dans : l’obésité à début précoce (avant 5 ans) et de caractère sévère (IMC>p99.5) accompagnée d’une hyperphagie(14). Les obésités avec des variants pathogènes bialléliques (LEP, LEPR, POMC, PCSK1 etc.) vont présenter une obésité beaucoup plus sévère que les formes monoalléliques (SH2B1, SIM1 etc.).
Les obésités monogéniques et syndromiques sont des maladies rares, avec une prévalence inférieure à 1 sur 2000 enfants. La demande d’analyses génétiques auprès de l’assurance maladie se fait par le biais d’un formulaire de maladie orpheline que nous pouvons retrouver sur le site de la Société Suisse de Génétique Médicale. L’analyse de gènes se fera par un exome ciblé (panel de gènes) ou une analyse de méthylation (SPW ou AHO) quand le phénotype est suggestif d’une maladie spécifique. Dans le cas complexes ou hétérogènes un avis génétique doit être demandé.
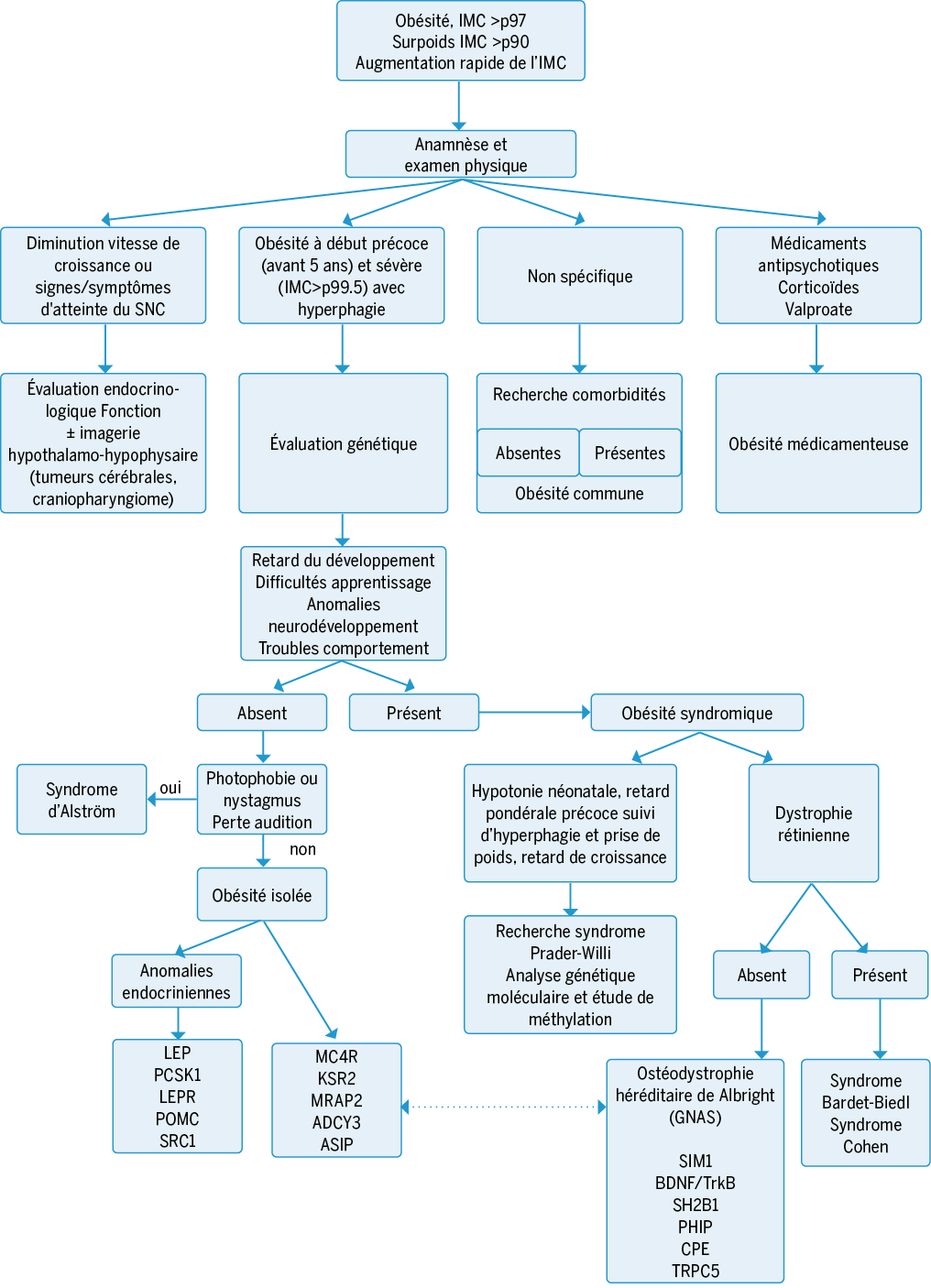
Prise en charge des obésités monogéniques et syndromiques(10)
Prise en charge multiprofessionnelle
Une prise en charge multiprofessionelle des obésités monogéniques et syndromiques est toujours nécessaire, impliquant des diététicien·ne·s, des spécialistes en activité physique adaptée et des endocrinologues pour suivre et traiter les anomalies endocriniennes associés ainsi que les complications métaboliques. Dans le cas des obésité syndromiques, les symptômes associés tels que les anomalies du comportement et du développement, mais également les trouble de la vision, les altérations de la fonction rénale etc. doivent faire l’objet d’un suivi multidisciplinaire coordonné y incluant une prise en charge psychologique / psychiatrique.
Thérapie pharmacologique
Dans le cas des obésités monogéniques, le diagnostic génétique permet d’envisager l’introduction d’un traitement ciblé
En cas d’obésité liée à un défaut sur la voie du MC4R (mutation du récepteur de MC4 ou de la leptine, de POMC, de PCSK1), le setmélanotide, un agoniste du MC4R, permet de rétablir la voie de signalisation. Ce médicament est autorisé dans ces indications dès l’âge de 6 ans dans l’Union européenne. Des essais cliniques sont en cours pour le déficit en leptine, mutation de MC4R, de SIM1, de SH2B1, et dans certaines obésités syndromiques : SA, BBS et SPW.
En cas de déficit congénital en leptine, le remplacement de l’hormone déficitaire (métréleptine), permet d’une part de corriger l’hyperphagie et ainsi de normaliser le poids, et de rétablir la fonction gonadotrope. Il faut toutefois rappeler le rôle procancereux de la leptine, ce qui cause une augmentation du risque de lymphome.
Analogues du GLP-1 (ou agonistes des récepteurs du GLP-1)
Le glucagon-like peptide-1 (GLP-1) est une hormone sécrétée par l’intestin au moment de la digestion, qui agit au niveau de récepteurs hypothalamiques (participe à la sensation de satiété) et au niveau digestif diminue la vidange gastrique et la motilité intestinale. Cela prolonge la satiété et diminue l’appétit. Par ailleurs le GLP-1 a un effet stimulateur de la sécrétion glucose-dépendante de l’insuline au niveau du pancréas et est utilisé pour le traitement du diabète de type 2.
Les analogues du GLP1 sont indiqués dans le traitement de l’obésité commune sévère, mais permettent également une perte de poids significative chez les patient·e·s souffrant de mutations de MC4R (voir article 6, Karen et al.).
Chirurgie bariatrique(20)
La chirurgie bariatrique permet en général une perte de poids chez les patient·e·s souffrant d’obésité monogénique ou syndromique, mais le suivi à long terme montre que la reprise de poids est plus importante et plus fréquente que chez les patient·e·s souffrant d’obésité commune, limitant l’intérêt à long terme de ce type de traitement. À ceci s’ajoute des complications chirurgicales et des réinterventions plus fréquentes chez les patient·e·s souffrant d’obésité syndromique, notamment chez les patient·e·s atteint·e·s de SPW. Les variantes hétérozygotes de la voie de la mélanocortine constituent une contre-indication relative à la chirurgie bariatrique(20).
Conclusions
La découverte des gènes impliqués dans l’obésité monogénique a permis de comprendre la voie de signalisation de la satiété et ainsi de proposer des thérapies ciblées efficace, notamment la métréleptine et le setmélanotide pour contrôler l’hyperphagie sévère et normaliser le poids. Le setmélanotide intervenant à la fin de la voie de signalisation, il est un bon candidat pour le traitement de certaines obésités syndromiques dont l’origine est hypothalamique (SPW, BBS, SA), et des essais cliniques sont en cours.
Références
- Fitch AK, Malhotra S, Conroy R. Differentiating monogenic and syndromic obesities from polygenic obesity: Assessment, diagnosis, and management. Obesity Pillars. 2024;11:100110. doi:10.1016/j.obpill.2024.100110
- Siddiqui J, Kinney CE, Han JC. The Genetics of Obesity. Pediatric Clinics of North America. 2024;71(5):897-917. doi:10.1016/j.pcl.2024.06.001
- Han JC, Rasmussen MC, Forte AR, Schrage SB, Zafar SK, Haqq AM. Management of Monogenic and Syndromic Obesity. Gastroenterology Clinics of North America. 2023;52(4):733-750. doi:10.1016/j.gtc.2023.08.005
- Berthoud HR, Münzberg H, Morrison CD. Blaming the Brain for Obesity: Integration of Hedonic and Homeostatic Mechanisms. Gastroenterology. 2017;152(7):1728-1738. doi:10.1053/j.gastro.2016.12.050
- Lister NB, Baur LA, Felix JF, et al. Child and adolescent obesity. Nat Rev Dis Primers. 2023;9(1):24. doi:10.1038/s41572-023-00435-4
- Han JC, Lawlor DA, Kimm SY. Childhood obesity. The Lancet. 2010;375(9727):1737-1748. doi:10.1016/S0140-6736(10)60171-7
- Berthoud HR, Morrison CD, Münzberg H. The obesity epidemic in the face of homeostatic body weight regulation: What went wrong and how can it be fixed? Physiology & Behavior. 2020;222:112959. doi:10.1016/j.physbeh.2020.112959
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289-295. doi:10.1038/nature05026
- Friedman JM. Leptin and the endocrine control of energy balance. Nat Metab. 2019;1(8):754-764. doi:10.1038/s42255-019-0095-y
- Hinney A, Körner A, Fischer-Posovszky P. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat Rev Endocrinol. 2022;18(10):623-637. doi:10.1038/s41574-022-00716-0
- Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. 2022;23(2):120-133. doi:10.1038/s41576-021-00414-z
- Zorn S, Von Schnurbein J, Schirmer M, Brandt S, Wabitsch M. Measuring hyperphagia in patients with monogenic and syndromic obesity. Appetite. 2022;178:106161. doi:10.1016/j.appet.2022.106161
- Farooqi IS. Monogenic Obesity Syndromes Provide Insights Into the Hypothalamic Regulation of Appetite and Associated Behaviors. Biological Psychiatry. 2022;91(10):856-859. doi:10.1016/j.biopsych.2022.01.018
- Styne DM, Arslanian SA, Connor EL, et al. Pediatric Obesity—Assessment, Treatment, and Prevention: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2017;102(3):709-757. doi:10.1210/jc.2016-2573
- Kohlsdorf K, Nunziata A, Funcke JB, et al. Early childhood BMI trajectories in monogenic obesity due to leptin, leptin receptor, and melanocortin 4 receptor deficiency. Int J Obes. 2018;42(9):1602-1609. doi:10.1038/s41366-018-0049-6
- Argente J, Farooqi IS, Chowen JA, et al. Hypothalamic obesity: from basic mechanisms to clinical perspectives. The Lancet Diabetes & Endocrinology. 2025;13(1):57-68. doi:10.1016/S2213-8587(24)00283-3
- Semenova E, Guo A, Liang H, Hernandez CJ, John EB, Thaker VV. The expanding landscape of genetic causes of obesity. Pediatr Res. Published online December 17, 2024. doi:10.1038/s41390-024-03780-6
- Tomlinson JW. Bardet‐Biedl syndrome: A focus on genetics, mechanisms and metabolic dysfunction. Diabetes Obesity Metabolism. 2024;26(S2):13-24. doi:10.1111/dom.15480
- Ruiz I, Bouthors T, Borloz S, Hauschild M, Maggio A, Schwitzgebel VM. Obésité infantile : nouveautés thérapeutiques ciblées. Revue Médicale Suisse. 2023;19(815):374-379. doi:10.53738/REVMED.2023.19.815.374
- Faccioli N, Poitou C, Clément K, Dubern B. Current Treatments for Patients with Genetic Obesity. Jcrpe. 2023;15(2):108-119. doi:10.4274/jcrpe.galenos.2023.2023-3-2
Informations complémentaires
Auteur·e·s
-
Dr med. Inge Lore Ruiz AranaUnité d’Endocrinologie, Diabétologie et Obésité, Département Femme-Mère-Enfant, CHUV, Lausanne
-
Dr med. Thérèse BouthorsUnité d’Endocrinologie, Diabétologie et Obésité, Département Femme-Mère-Enfant, CHUV, Lausanne
-
Dr med. Maria-Christina AntoniouUnité d’Endocrinologie, Diabétologie et Obésité, Département Femme-Mère-Enfant, CHUV, Lausanne