Abstract
Le syndrome hémolytique et urémique (SHU) est une des causes les plus fréquentes d’insuffisance rénale aiguë nécessitant une dialyse pendant l’enfance. Le SHU est une maladie multi-systémique qui peut occasionner des manifestations et complications rénales et extrarénales aiguës et à long terme. Du point de vue étiologique on distingue les formes fréquentes (env. 90-95%) associées à une infection ainsi que les rares (env. 5-10%) activées par le complément et d’autres. Le traitement et le pronostic étant fondamentalement distincts entre les différentes formes de SHU, un diagnostic étiologique minutieux est indispensable. Les patients avec un SHU nécessitent un suivi néphrologique à long terme afin de détecter et traiter précocement de possibles complications rénales.
Introduction
Le syndrome hémolytique et urémique (SHU) est une des causes les plus fréquentes d’insuffisance rénale aiguë nécessitant une dialyse pendant l’enfance. En Suisse chaque année environ 1.4/100’000 enfants de ≤16 ans contractent un SHU1). Le SHU est une maladie multi-systémique pouvant être associée à des manifestations rénales et extrarénales potentiellement létales2-6). Grâce à l’amélioration des moyens de surveillance et de traitement intensifs ainsi qu’une compréhension physiopathologique globale des différentes formes de SHU pendant les derniers 60 ans, la mortalité pendant l’enfance est tombée, selon les formes de SHU, d’env. 40-50% à env. 1-12%. 1,2,7)
Définition
Le SHU se définit par la triade
- anémie hémolytique non-immunologique avec mise en évidence de fragmentocytes dans le frottis de sang périphérique
- thrombocytopénie resp. signes de déplétion thrombocytaire et
- signes d’une atteinte rénale aiguë (oligurie/anurie, créatininémie élevée, protéinurie, hématurie, anomalies rénales à l’échographie),
dont l’origine histopathologique est une microangiopathie thrombotique (MAT)1). Du point de vue physiopathologique la MAT se caractérise par une atteinte endothéliale avec formation de microthrombi dans les petits vaisseaux8).
Classification basée sur l’étiologie, physiopathologie et épidémiologie
Généralités
La triade décrite n’est pas spécifique au SHU mais se rencontre aussi dans d’autres formes de MAT. En raison des changements de concepts physiopathologiques, la classification des MAT resp. SHU a subi ces dernières décennies de nombreuses mutations. La distinction utilisée longtemps en formes SHU avec ou sans diarrhée ou typiques et atypiques a cédé la place à une classification détaillée basée sur l’étiologie3,8,9) (figure 1).

D’après Loirat et al. 2016 9)
Abréviations: ADAMTS13, A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; CFB, facteur B du complément; CFH, facteur H du complément; CFI, facteur I du complément; DGKE, diacylglycérolkinase ε; HELLP, Hemolysis, Elevated Liver Enzymes, And Low Platelet Count Syndrome; VIH, Virus d’immunodéficience humaine; MCP, Membrane Cofactor Protein (CD46); STEC, Escherichia coli produisant des Shigatoxines.
Pendant l’enfance on rencontre surtout les formes de SHU associées à une infection (env. 90-95%, les formes par activation du complément et d’autres formes congénitales ou secondaires de SHU étant nettement plus rares (env. 5-10%)2). Parmi les formes de MAT, le diagnostic différentiel du SHU le plus important, néanmoins très rare chez l’enfant et l’adolescent, est le purpura thrombotique thrombocytopénique (PTT). L’âge de la première manifestation peut donner des indications importantes sur l’étiologie et on doit absolument en tenir compte en établissant le diagnostic différentiel d’une MAT ou SHU (figure 2).
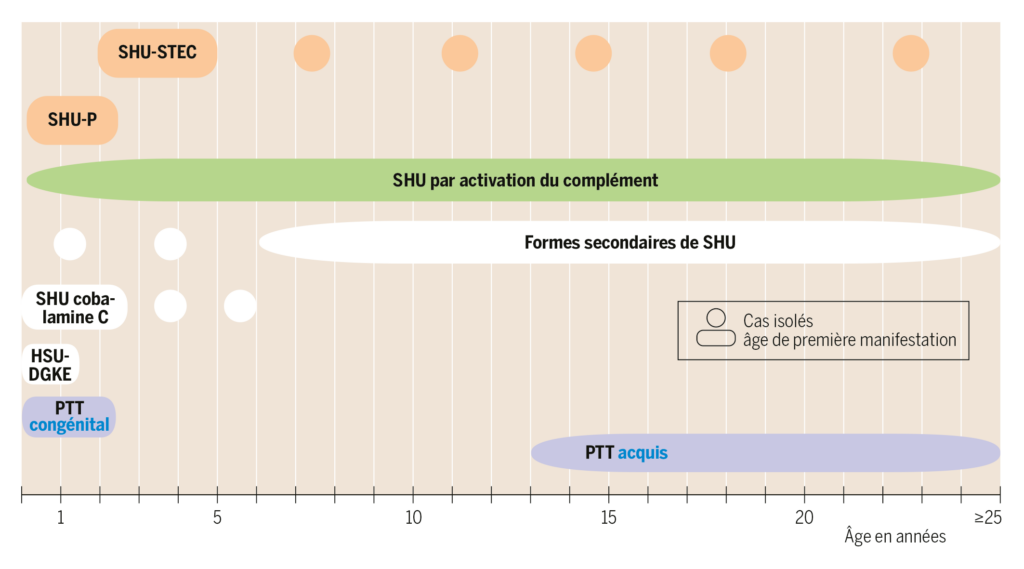
D’après Holle J et al. 2017, S.1009 3)
Abréviations: DGKE, diacylglycérolkinase ε; SHU, syndrome hémolytique et urémique; SHU-P, SHU associé à Streptococcus pneumoniae; STEC, Escherichia coli produisant la Shigatoxine; PTT, purpura thrombotique-thrombocytopénique
SHU par des germes produisant la Shigatoxine (SHU-STEC)
Le SHU associé aux Shigatoxines représente avec env. 90% la forme la plus fréquente de SHU pendant l’enfance. La cause est une infection par des germes produisant des Shigatoxines, le plus souvent E. coli entéro-hémorragiques (EHEC) ou plus rarement aussi Shigella dysenteriae type 1, dont les toxines libérées endommagent l’endothélium et occasionnent la MAT2,8). Les sources d’infections EHEC sont les produits laitiers non pasteurisés crus, les aliments contaminés et le contact avec des animaux. Néanmoins, seuls 5-15% environ de tous les patients avec une infection EHEC développent, dans les 7 à 14 jours après le début des symptômes gastro-intestinaux, un SHU-STEC2,3), affection qui s’observe typiquement chez des enfants âgés 2 à 5 ans3) (figure 2). Notamment dans le cadre d’une situation épidémique, d’autres tranches d’âge peuvent être concernées10).
SHU associé à Streptococcus pneumoniae (SHU-P)
Les infections invasives à Streptococcus pneumoniae peuvent également déclencher dans 0.4-0,6% des cas, après un temps de latence de 3-18 jours, un SHU (SHU-P)2,7). La N-acétyl-neuraminidase libérée par les pneumocoques expose, chez les patients atteints, l’antigène Thomsen-Friedenreich à la surface des érythrocytes, thrombocytes et cellules endothéliales glomérulaires. Par la fixation d’anticorps IgM préformés à l’antigène Thomsen-Friedenreich, se produit une poly-agglutination et, par des mécanismes à ce jour pas entièrement compris, le développement d’une MAT7,8). Le SHU-P est désigné, à l’instar du SHU-STEC, comme étant un SHU associé à une infection; il représente env. 5% des SHU pédiatriques et 40-50% des cas SHU non-STEC7,11). Il apparait essentiellement chez les nourrissons et petits enfants de <2 ans7) (figure 2).
SHU par activation du complément
Avec une incidence d’env. 2:1’000’000 personnes et 5-10% de tous les cas SHU pédiatriques, les formes de SHU par activation du complément sont également rares3). À l’origine se trouvent différents défauts héréditaires autosomiques récessifs ou autosomiques dominants d’éléments du système du complément et/ou des auto-anticorps H acquis8,9) (figure 1). Actuellement l’identification de la/des mutation(s) concernée(s) dans les gènes codant les composants du système du complément, n’est possible que chez 60-70% des patients atteints9). Ces défauts du complément, stimulés par des facteurs endo- ou exogènes (p.ex. infections, opérations, grossesse) endommagent, par une activation incontrôlée de la voie alterne du complément, l’endothélium et occasionnent des MAT8,9). Le SHU par activation du complément se manifeste à tout âge et, contrairement aux formes de SHU associés aux infections mentionnées, surtout de façon récidivante12) (figure 2).
Autres formes de SHU
Outre des formes de SHU congénitales (déficit en cobalamine C, mutation de la diacylglycérolkinase Ɛ) se manifestant chez les nourrissons et petits enfants, peuvent apparaître, chez l’enfant et l’adulte, différentes formes secondaires de SHU suite à la prise de certains médicaments (p.ex. inhibiteurs de la calcineurine), après transplantation d’organes ou de cellules souches et/ou dans le cadre de maladies sous-jacentes les plus diverses (p.ex. lupus systémique ou pancréatite)8,9) (figure 1).
Purpura thrombotique thrombopénique (PTT)
Le PTT, dû également à une MAT, est associé à une activité réduite à <10% de la protéase ADAMTS13 clivant le facteur von Willebrand (A Disintegrin-like And Metalloprotease with a ThromboSpondin type 1 motif, member 13). Elle peut être d’origine congénitale (syndrome de Upshaw-Schulman) ou auto-immune (présence d’anticorps anti-ADAMTS13) (figure 1). À cause de l’activité réduite de cette protéase, la scission des multimères du facteur von Willebrand n’a pas lieu. L’accumulation de ces multimères induit une agrégation des thrombocytes dans la microcirculation et donc la MAT13). La maladie est très rare et il n’existe pas de chiffres fiables sur l’incidence pendant l’enfance et l’adolescence.
Anamnèse et clinique
Chez de nombreux patients avec un SHU on observe une détérioration de l’état général en peu de jours. Typiques sont les symptômes suivants:
1. pâleur, fatigue, inappétence et diminution de l’absorption de liquide et/ou diminution de l’activité physique
2. éventuellement pétéchies, saignements
3. diminution du volume urinaire jusqu’à l’anurie; év. œdèmes, occasionnellement macrohématurie.
Ces symptômes peuvent être isolés ou combinés.
Environ 40-75 (-85)% des patients développent une insuffisance rénale aiguë nécessitant une dialyse1,2,10,11). Les autres symptômes cliniques dépendent de l’étiologie et d’éventuelles manifestations extrarénales.
SHU-STEC
Les enfants avec un SHU-STEC présentent typiquement des symptômes gastro-intestinaux, en général nausées, vomissements et diarrhée (souvent sanguinolente). Cette symptomatologie est plus ou moins marquée, allant d’une quasi absence de symptômes à des évolutions sévères avec des complications gastro-intestinales tels des colites ischémiques et perforations intestinales2,10). Par rapport aux possibles situations endémiques ou épidémiques de SHU, il est important d’élargir l’anamnèse à l’entourage (famille, crèche, école, etc.), en recherchant d’autres personnes avec une symptomatologie semblable.
SHU-P
Les symptômes du SHU-P sont dictés surtout par l’infection à pneumocoques préalable. Celle-ci se présente en général sous forme de pneumonie (70%) ou de méningo-encéphalite (20-30%) et plus rarement de bactériémie à Streptococcus pneumoniae, sinusite ou otite moyenne7,10).
SHU par activation du complément
Le SHU par activation du complément aussi est souvent précédé par une infection, dans environ un tiers des cas par une gastro-entérite9). Un âge « atypique » pour la manifestation d’un SHU-STEC ou -P (<1 an, >5 ans), l’absence de symptômes d’une éventuelle infection à EHEC ou pneumocoques, une anamnèse familiale mentionnant d’autres personnes atteintes de SHU et notamment des épisodes de SHU récidivants sont des indices pour un SHU par activation du complément. Ces épisodes peuvent aller d’une évolution bénigne avec hémolyse modérée jusqu’à la forme sévère du SHU avec complications rénales et extrarénales9).
Autres formes rares de SHU
En raison de la multitude de causes congénitales ou acquises, les autres formes de SHU ne se laissent pas décrire par des symptômes caractéristiques (figure 1). Essentiel pour ces formes de SHU est la prise en compte de l’âge ainsi que des maladies préexistantes, de médicaments pris à long terme et de l’anamnèse familiale (figure 2).
Purpura thrombotique thrombocytopénique (PTT)
La clinique et l’anamnèse ne laissent pas clairement différencier le PTT des formes de SHU par activation du complément, le PTT pouvant aussi être déclenché par une infection et évoluer par poussées13). Comparé au SHU, le PTT a tendance à développer des manifestations rénales plus modérées et des complications neurologiques plus fréquentes13).
SHU – une maladie multi-organique
Indépendamment de l’étiologie, le SHU n’est pas une maladie exclusivement rénale mais multi-systémique, pouvant être associée à des complications extrarénales mettant la vie en danger2,3,9). Les symptômes extrarénaux possibles sont présentés dans le tableau 1. L’implication du système nerveux central représente avec 17-29% la manifestation extrarénale et en même temps la cause de décès la plus fréquente due au SHU1,2,4,5,6, 12,14).

Diagnostic
Généralités
À l’aide de la formule sanguine, des paramètres de rétention urinaire et hémolyse et des indications anamnestiques, il est dans la plupart des cas possible de poser le diagnostic de SHU en quelques heures.
Chez tout patient avec un SHU resp. MAT des examens complémentaires sont impératifs afin de:
1. évaluer l’extension des manifestations rénales et extrarénales et entreprendre les mesures nécessaires
2. pouvoir mettre en route précocement un traitement spécifique
3. pouvoir évaluer l’évolution et le pronostic et pouvoir planifier le suivi.
Le tableau 2 résume les examens biologiques et techniques permettant d’évaluer les manifestations rénales et extrarénales et de préciser l’étiologie. Ces examens peuvent et devraient être adaptés, réduits ou élargis en tenant compte des caractéristiques individuelles du patient.

En présence d’une anamnèse claire, d’un âge « typique », d’une anamnèse familiale sans particularités, d’un germe « typique » (EHEC ou pneumocoques) et donc d’un diagnostic évident d’un SHU-STEC ou –P, on pourra (dans un premier temps) renoncer à d’ultérieurs examens complémentaires.
Devant toute divergence de l’anamnèse, âge ou évolution « typiques », p.ex. atteinte rénale sévère sans récupération de la fonction glomérulaire, atteinte neurologique ou cardiologique sévères, anamnèse familiale positive, absence de mise en évidence du germe et surtout évolution récidivante, l’exclusion des autres formes de SHU et de PTT par des examens plus approfondis est impérative (figure 2 et tableau 2).
Diagnostic du système du complément
L’interprétation des examens concernant le complément, servant surtout à différentier les SHU par activation du complément des autres formes SHU resp. MAT, peut s’avérer difficile dans le cas individuel. La mise en évidence d’un taux plasmatique réduit de C3 (et év. C4) parle en principe en faveur d’une consommation de complément et donc d’une activation du complément. Par contre, pas tous les patients avec un SHU par activation du complément présentent une hypocomplémentémie7,9). Pour une meilleure appréciation de l’étendue de l’activation du complément, des examens spécifiques avec dosage de divers facteurs du système du complément, des auto-anticorps contre certains facteurs du complément ainsi que la concentration du complexe terminal du complément s’avèrent nécessaires (tabl. 2).
Diagnostic d’ADAMTS13
En constatant une activité ADAMTS13 de <10% on doit admettre un PTT. La différenciation entre un PTT congénital ou acquis est possible par l’âge et la mise en évidence des anticorps ADAMTS13 (fig. 1).
Importance et cadre temporel du diagnostic
Il est impératif, tant pour le traitement que pour le pronostic, de procéder rapidement à un diagnostic différentiel et de déterminer l’étiologie d’un SHU ou d’un PTT. Le problème réside dans le fait qu’actuellement de nombreux examens indispensables durent des semaines voire des mois, alors que, pour éviter des séquelles irréversibles rénales et extrarénales, les mesures thérapeutiques doivent être prises rapidement et la plupart des fois avant de connaître le diagnostic définitif.
Traitement
Traitement de base pour toutes les formes de SHU
La prise en charge médicale des enfants et adolescents avec un SHU ou MAT devrait se faire dans un centre de néphrologie pédiatrique ou en collaboration avec un néphrologue pédiatre. En principe, et surtout au début, est nécessaire une surveillance étroite, souvent dans un service de soins intensifs pédiatriques, des paramètres cardio-respiratoires et neurologiques, de l’excrétion urinaire et des paramètres biologiques essentiels (formule sanguine, indicateurs d’hémolyse et de rétention urinaire, électrolytes, équilibre acidobasique. En raison des complications et évolutions potentiellement létales, les patients devraient être transférés tôt dans un centre spécialisé, disposant de toutes les possibilités de surveillance et traitement de soins intensifs pédiatriques (y compris les différentes formes de thérapie de remplacement rénal et d’aphérèse).
Le traitement de soutien de toutes les formes de SHU ou MAT consiste en un apport liquidien équilibré, un volume adéquat en cas de déshydratation, le traitement médicamenteux et/ou diététique en cas de complications rénales secondaires (acidose métabolique, hyperkaliémie, hyperphosphatémie), un apport calorique suffisant pour éviter un catabolisme, ainsi que la transfusion (restrictive) de produits sanguins (p.ex. concentré d’érythrocytes pour le patient instable sur le plan hémodynamique, concentré de plaquettes en cas de saignements et év. avant une intervention chirurgicale)2,3,8).
Si l’indication à la dialyse est posée, on procède généralement, surtout chez les petits enfants, d’abord à une dialyse péritonéale; en cas de contre-indication à la dialyse péritonéale (p.ex. colite hémorragique sévère ou perforation intestinale suite à un SHU-STEC) ou chez les enfants plus âgés et adolescents, on pratique des techniques de filtration du sang extracorporelles, comme l’hémodialyse continue ou intermittente ou l’hémo(dia)filtration.
SHU-STEC
Il n’existe actuellement de traitement causal du SHU-STEC. Une amélioration jusqu’à une guérison totale a été obtenue, dans certains cas avec des complications neurologiques sévères, par plasmaphérèse ou inhibition du complément C5 par éculizumab; des recommandations thérapeutiques claires manquent néanmoins à l’heure actuelle2,10,14).
SHU-P
Les infections invasives à pneumocoques nécessitent un traitement antibiotique. Pour le SHU-P n’existe, comme pour le SHU-STEC, pas de traitement causal.
Traitement spécifique du SHU par activation du complément
Lorsque la preuve d’un SHU associé à une infection, y compris SHU-STEC et SHU-P (voir ci-dessus) fait défaut, et après avoir exclu un PTT on admettra, en raison des conséquences thérapeutiques et jusqu’à preuve du contraire, un SHU par activation du complément. Le traitement de choix, initié immédiatement et avant d’avoir obtenu les résultats biologiques définitifs, pour un SHU par activation du complément consiste en l’administration d’anticorps monoclonaux anti-C5 éculizumab (Soliris®)8,9). En bloquant la fraction C5 du complément, éculizumab interrompt les réactions secondaires de la cascade et la formation du complexe terminal du complément, empêchant ainsi l’atteinte endothéliale et la MAT. Après des doses initiales plus rapprochées, l’administration d’éculizumab se fait, à partir de 3 semaines, tous les 14 jours par perfusion intraveineuse. Parmi les effets indésirables il faut surtout mentionner la sensibilité accrue vis-à-vis des bactéries encapsulées, notamment Neisseria meningitidis (avec un risque relatif de 5’000 comparé à la population normale)8,9). En raison de l’évolution récidivante des formes de SHU par activation du complément, on recommande actuellement un traitement à vie par blocage du système du complément, bien que des descriptions de cas isolés ou de petites séries rapportent des traitements interrompus, en fonction de facteurs de risque individuels, avec succès15). Plusieurs inhibiteurs du complément alternatifs sont actuellement en voie de développement.
Traitement spécifique d’autres formes de SHU
Le traitement spécifique des autres formes de SHU dépend de la maladie sous-jacente (p.ex. substitution d’hydroxycobalamine et folate pour le SHU dû à un déficit en cobalamine; traitement immunosuppresseur d’un lupus systémique; interruption de traitements médicamenteux occasionnant un SHU ou une MAT (p.ex. inhibiteurs de la calcineurine)2,8).
PTT
Le PTT congénital est traité par perfusions de plasma; les ADAMTS13 contenus dans le plasma empêchent l’atteinte endothéliale et la MAT13). Pour les PTT acquis l’élimination des anticorps anti-AMATS13 par plasmaphérèse représente le traitement de choix; lors d’une évolution récidivante on peut envisager une immunomodulation par déplétion des cellules B au moyen de l’anticorps rituximab dirigé contre l’antigène de surface CD2013).
Pronostics, effets à long terme et suivi
Globalement le pronostic à long terme après un SHU est bon.
Pronostic rénal
Seulement environ 1-5% des patients pédiatriques avec un SHU-STEC ou SHU-P développent une insuffisance rénale terminale chronique nécessitant une thérapie de substitution rénale1,10,14). Une partie non négligeable d’environ 25-30% des patients avec un SHU-STEC et SHU-P développe néanmoins des troubles rénaux persistants ou apparaissant plus tard (protéinurie, hypertension et/ou diminution de la fonction glomérulaire)1,3,11,12,14). À l’heure actuelle on ne dispose que de peu de données concernant le devenir rénal, à l’âge adulte, après un SHU associé à une infection pendant l’enfance. En raison des possibles complications à long terme, le suivi néphrologique est vivement recommandé tant pendant l’enfance qu’à l’âge adulte1-3,9,14).
Le pronostic des formes de SHU par activation du complément a changé significativement depuis que s’est établi le traitement par inhibition du complément: avant son introduction et en fonction du type de déficit du complément, dans jusqu’à 90% des cas l’évolution était récidivante, jusqu’à 83% des cas évoluaient vers une insuffisance rénale terminale, et après transplantation rénale allogène jusqu’à 90% subissaient une récidive de la maladie originale avec perte de la greffe2,3,12). Entre temps, pour la plupart des formes de SHU par activation du complément les récidives, l’insuffisance rénale terminale et la récidive après une greffe du rein peuvent être évitées ou traitées.
En raison des étiologies très variées des autres formes de SHU, des indications générales concernant le devenir, conditionné par la maladie sous-jacente, ne sont pas possibles.
Pronostic extrarénal
Les manifestations extrarénales particulièrement sévères pendant la phase aiguë d’un SHU peuvent engendrer, indépendamment de l’étiologie du SHU, des complications extrarénales à long terme, p.ex. sous forme de pathologies neurologiques persistantes lors d’une implication du système nerveux central, de diabète suite à une pancréatite ou de troubles persistants de la vue dus à une atteinte de la rétine1,10,11,14).
Conclusion
- Le diagnostic de SHU est rapidement possible à partir de la triade typique.
- La clarification de l’étiologie du SHU comprend une anamnèse minutieuse et un diagnostic différentiel approfondi, indispensable aux traitements spécifiques et à l’établissement d’un pronostic.
- Pour tout SHU un suivi régulier est fortement recommandé, indépendamment de l’étiologie, aussi à l’âge adulte.
Littérature
- Schifferli A, von Vigier RO, Fontana M, Spartà G, Schmid H, Bianchetti MG, et al. Hemolytic-uremic syndrome in Switzerland: a nationwide surveillance 1997-2003. Eur J Pediatr. 2010;169(5):591-8.
- Loirat C, Saland J, Bitzan M. Management of hemolytic uremic syndrome. Presse Med. 2012;41(3 Pt 2):e115-e35.
- Holle J, Lange-Sperandio B, Mache C, Oh J, Pape L, Schaefer F, et al. Hämolytisch-urämisches Syndrom im Kindes- und Jugendalter. Monatsschr Kinderheilkd. 2017;165:1005-18.
- Matthies J, Hunseler C, Ehren R, Volland R, Korber F, Hoppe B, et al. Extrarenal Manifestations in Shigatoxin-associated Haemolytic Uremic Syndrome. Klin Padiatr. 2016;228(4):181-8.
- Siegler RL. Spectrum of extrarenal involvement in postdiarrheal hemolytic-uremic syndrome. J Pediatr. 1994;125(4):511-8.
- Hofer J, Rosales A, Fischer C, Giner T. Extra-renal manifestations of complement-mediated thrombotic microangiopathies. Front Pediatr. 2014;2:97.
- Scobell RR, Kaplan BS, Copelovitch L. New insights into the pathogenesis of Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol. 2020;35(9):1585-91.
- Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. 2017;390(10095):681-96.
- Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15-39.
- Loos S, Ahlenstiel T, Kranz B, Staude H, Pape L, Hartel C, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-9.
- Waters AM, Kerecuk L, Luk D, Haq MR, Fitzpatrick MM, Gilbert RD, et al. Hemolytic uremic syndrome associated with invasive pneumococcal disease: the United kingdom experience. J Pediatr. 2007;151(2):140-4.
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey M-A, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-62.
- Loirat C, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura in children. Curr Opin Pediatr. 2013;25(2):216-24.
- Rosales A, Hofer J, Zimmerhackl L-B, Jungraithmayr TC, Riedl M, Giner T, et al. Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin Infect Dis. 2012;54(10):1413-21.
- Fakhouri F, Fila M, Provot F, Delmas Y, Barbet C, Chatelet V, et al. Pathogenic Variants in Complement Genes and Risk of Atypical Hemolytic Uremic Syndrome Relapse after Eculizumab Discontinuation. Clin J Am Soc Nephrol. 2017;12(1):50-9.
Informations complémentaires
Auteur·e·s
-
Dr. med. Kathrin BuderAbteilung Pädiatrische Nephrologie, Universitäts-Kinderspital Zürich – Eleonorenstiftung
-
Dr. med. Markus FeldkötterAbteilung Pädiatrische Nephrologie, Universitäts-Kinderspital Zürich – Eleonorenstiftung
-
PD Dr. med. Giuseppina SpartàAbteilung Pädiatrische Nephrologie, Universitäts-Kinderspital Zürich – Eleonorenstiftung